Eudamedin julkisen rekisterin tarkoituksena on tarjota kansalaisille mahdollisuus saada riittävästi tietoa EU:n markkinoilla olevista lääkinnällisistä laitteista.
Julkinen rekisteri kattaa tiedot:
asianomaisista talouden toimijoista (valmistaja, valtuutettu edustaja, järjestelmän tai toimenpidepakkauksen kokoaja, maahantuoja)
markkinoilla olevista lääkinnällisistä laitteista ja UDI-tietokannan
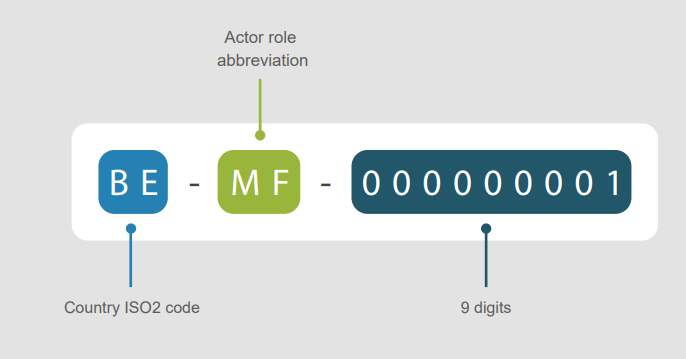
Toimijamoduuli luo rekisteröityneille toimijoille yksilöllisen tunnisteen. Tunnistetta kutsutaan SRN-tunnisteeksi, jos toimija rekisteröityy asetuksen (EU/2017/745 tai EU/2017/746) mukaisesti. Muussa tapauksessa tunnistetta kutsutaan actor ID:ksi.
Tunniste myönnetään, kun toimintavaltainen viranomainen on vahvistanut rekisteröintipyynnön. Tunniste muodostuu maakoodista, toimijan roolista ja 9 numerosta.
Toimijat voivat jo syöttää tiedot EUDAMEDiin, mutta Euroopan komissio ei voi vaatia toimijamoduulin käyttöä ennen kuin EUDAMED on täysin toimintakykyinen asetuksien mukaisesti.
Kuitenkin asetuksen siirtymäsäännöt antavat viranomaisille valtuudet päättää rekisteröinneistä jäsenvaltiokohtaisesti ennen kuin EUDAMED on täysin toimintakykyinen. Suomessa toimijoiden ja laitteiden rekisteröinti tapahtuu EUDAMED- tai CERE-rekisteriin.
EUDAMEDista löytyy kotimaiset lääkinnällisen laitteen valmistajat, maahantuojat, valtuutetut edustajat, järjestelmän tai toimenpidepakkauksen kokoajat tai näiden steriloijat ja yksilölliseen käyttöön tarkoitetun laitteen valmistajat (mikäli laitteen vaatimustenmukaisuuden arvioinnissa on käytetty ilmoitettua laitosta). Loput toimijoista ovat CERE:ssä, esimerkiksi jakelijat.
Laitteisiin on otettu käyttöön yksilöllinen laitetunnisteita koskeva järjestelmä (UDI), joka helpottaa laitteiden tunnistamista ja jäljitettävyyttä. UDI-tunniste voi olla viivakoodi, QR-koodi tai mikä tahansa muu koneluettava koodi. UDI-tunnisteet lisätään merkintöihin laitteen riskiluokan mukaan vaiheittain vuoteen 2027 mennessä.
Laitteiden tunnistaminen ja jäljitettävyys edellyttää, että kaikki EU:n markkinoilla olevat valmistajat toimittavat laitteiden UDI-/laitetiedot EUDAMEDiin. Valmistajat voivat jo syöttää tiedot EUDAMEDiin, mutta Euroopan komissio ei voi vaatia moduulin käyttöä ennen kuin EUDAMED on täysin toimintakykyinen asetuksien mukaisesti.
Kuitenkin asetuksen siirtymäsäännöt antavat viranomaisille valtuudet päättää rekisteröinneistä jäsenvaltiokohtaisesti ennen kuin EUDAMED on täysin toimintakykyinen. Suomessa toimijoiden ja laitteiden rekisteröinti tapahtuu EUDAMED- tai CERE-rekisteriin.
Ilmoitettujen laitosten on rekisteröitävä EUDAMESiin kaikki tiedot myönnetyistä, keskeytetyistä, palautetuista, perutuista tai evätyistä sertifikaateista. Ilmoitetut laitokset voivat jo syöttää tiedot EUDAMEDiin, mutta Euroopan komissio ei voi vaatia moduulin pakollista käyttöä ennen kuin EUDAMED on täysin toimintakykyinen asetuksien mukaisesti.
Ilmoitetut laitokset
Ilmoitetut laitokset ovat EU:n jäsenvaltioiden nimeämiä riippumattomia ja puolueettomia vaatimustenmukaisuuden arviointilaitoksia. Lääkinnällisten laitteiden asetusten (EU) 2017/745 ja (EU) 2017/746 mukainen vaatimustenmukaisuuden arviointi edellyttää ilmoitetun laitoksen osallistumista arviointiin ennen korkean riskin laitteiden markkinoille saattamista. Ilmoitettu laitos arvioi laitteen ja auditoi valmistajan toiminnan. Vaatimusten täyttymisisestä myönnetään valmistajalle todistus.
Kussakin EU-jäsenmaassa on ilmoitettujen laitosten nimeämisestä ja valvonnasta vastaava viranomainen. Suomessa Fimea vastaa ilmoitettujen laitosten MD- ja IVD-asetusten mukaisesta nimeämisestä ja valvonnasta. Luettelo ilmoitetuista laitoksista ja niiden pätevyysalueista löytyy EU:n komission ylläpitämästä NANDO-rekisteristä. Suomeen sijoittuneita, MD-asetuksen mukaisia ilmoitettuja laitoksia on tällä hetkellä kaksi, Eurofins Electric & Electronics ja SGS Fimko Oy.
Kliiniset tutkimukset ja suorituskyvyn arviointi moduuli ei ole vielä käytössä. Moduulissa on tulevaisuudessa mahdollisuus päästä mm. tutkimusraporttien yhteenvetoihin ja kliinistä suorituskykyä koskeviin raportteihin
Vaaratilanteiden ja markkinoille saattamisen jälkeinen valvonta moduuli ei ole vielä käytössä. Moduulissa on tulevaisuudessa mahdollisuus päästä mm. osittaisen pääsyn valmistajan vaaratilanneilmoitusraportteihin (manufacturer incident reports) sekä kenttäturvallisuusilmoituksiin (field safety notices)
Markkinavalvonta moduuli ei ole vielä käytössä. Moduulissa on tulevaisuudessa mahdollisuus päästä mm. kunkin jäsenvaltion yhteenvetoon kansallisen alueen markkinavalvontatoimien tuloksista.
Euroopan komissio ei voi vaatia toimija- tai UDI-/laitemoduulin käyttöä ennen kuin Eudamed on täysin toimintakykyinen asetuksien mukaisesti. Lisäksi asetuksissa on määritelty siirtymäajat, milloin kaikkien EU:n jäsenvaltioiden on otettava Eudamed käyttöön. Nämä siirtymäajat alkavat kulumaan vasta, kun Eudamed on täysin toimintakykyinen. Täysin toimintakykyisellä tarkoitetaan asetuksen mukaan sitä, kun kaikki Eudamedin moduulit ovat käytössä.
Kuitenkin asetuksen siirtymäsäännöt antavat viranomaisille valtuudet päättää rekisteröinneistä jäsenvaltio kohtaisesti ennen kuin Eudamed on täysin toimintakykyinen. Suomessa toimijoiden ja laitteiden rekisteröinti tapahtuu EUDAMED- tai CERE-rekisteriin.
Eudamedista löytyy kotimaiset lääkinnällisen laitteen valmistajat, maahantuojat, valtuutetut edustajat, järjestelmän tai toimenpidepakkauksen kokoajat tai näiden steriloijat ja yksilölliseen käyttöön tarkoitetun laitteen valmistajat (mikäli laitteen vaatimustenmukaisuuden arvioinnissa on käytetty ilmoitettua laitosta). Loput toimijoista ovat CERE:ssä, esimerkiksi jakelijat.
EUDAMED-rekisteri on komission ylläpitämä rekisteri. Tulevaisuudessa kaikkien EU:n jäsenvaltioiden on otettava EUDAMED käyttöön, mutta tällä hetkellä EUDAMED-rekisterin käyttö EU:ssa on vapaaehtoista. Asetuksissa (EU/2017/745 ja EU/2017/746) on määritelty pitkä siirtymäajat sille, milloin kaikkien EU:n jäsenvaltioiden on otettava Eudamed käyttöön. Tästä johtuen EUDAMED ei ole vielä kaikissa jäsenvaltioissa käytössä.
Suomessa EUDAMED-rekisterin on otettu käyttöön, joten jokainen Suomessa sijaitseva lääkinnällisen laitteen valmistaja, maahantuoja, valtuutettu edustaja, järjestelmän tai toimenpidepakkauksen kokoaja tai näiden steriloija ja yksilölliseen käyttöön tarkoitetun laitteen valmistaja (mikäli laitteen vaatimustenmukaisuuden arvioinnissa on käytetty ilmoitettua laitosta) löytyy EUDAMED-rekisteristä.
Suomessa EU-säätely, kansallinen laki lääkinnällisistä laitteista 719/2021 sekä Fimean määräys 2/2021 määrittelee, ketkä lääkinnällisten laitteiden toimijoista ilmoittavat laite- ja toimijatietonsa EUDAMED-rekisteriin. Osa lääkinnällisten laitteiden toimijoista ilmoittaa tietonsa CERE-rekisteriin.