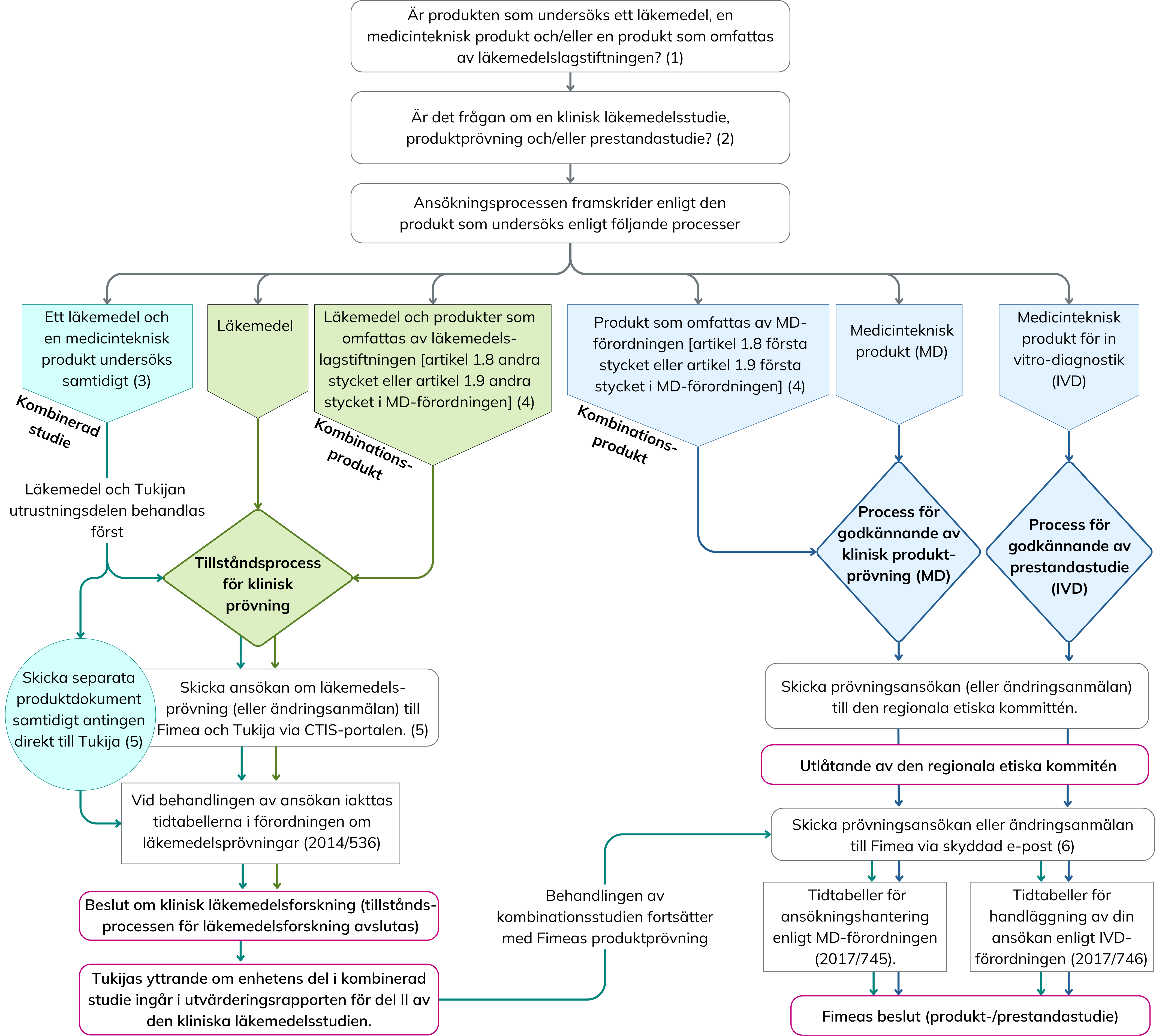
Kombinationsprövningar
Om en klinisk prövning för en medicinteknisk produkt eller en prestandastudie för en IVD-produkt ska utföras på så sätt att den kombineras med en klinisk läkemedelsprövning, ska följande beaktas vid planeringen av prövningen:
- Vid kombinationsprövningar gäller lagstiftningen om båda. Prövningens läkemedelsdel bedöms av läkemedelsmyndigheten enligt förordning (EU) 536/2014. Prövningens produktdel bedöms av produktmyndigheten enligt MD- eller IVD-förordningen.
- Vid kombinationsprövningar utförs den etiska utvärderingen av Nationella kommittén för medicinsk forskningsetik (Tukija).
- I nuläget finns det inte en harmoniserad process för myndighetsbehandlingen av ansökningarna, och därför ska prövningen planeras i enlighet med lagstiftningen för båda.
Enligt lagstiftningen för medicintekniska produkter förutsätts ett utlåtande av etiska kommittén innan prövningen lämnas för bedömning av myndigheten. Men i läkemedelslagstiftningen framskrider etiska kommitténs utvärdering samtidigt som myndighetens bedömning. Därför utförs bedömningen av läkemedelsprövningen vanligen först (tillsammans med etiska kommitténs utvärdering), och därefter lämnas prövningen för bedömning av produktmyndigheten.